
Sisu
- Põhjused
- Sümptomid
- Eksamid ja testid
- Ravi
- Tugirühmad
- Outlook (prognoos)
- Millal pöörduda arsti poole
- Ärahoidmine
- Alternatiivsed nimed
- Pildid
- Viited
- Läbivaatamise kuupäev 10/15/2018
Niemann-Picki haigus (NPD) on rühm haigusi, mis on möödunud perede kaudu (päritud), kus rasvained, mida nimetatakse lipiidideks, kogunevad põrna, maksa ja aju rakkudesse.
Haigus on kolm levinud vormi:
- Tüüp A
- Tüüp B
- Tüüp C
Iga tüüp hõlmab erinevaid organeid. See võib hõlmata närvisüsteemi ja hingamist. Igaüks võib põhjustada erinevaid sümptomeid ja võib esineda erinevatel aegadel kogu elu jooksul.
Põhjused
NPD tüübid A ja B tekivad siis, kui kehas olevatel rakkudel ei ole ensüümi, mida nimetatakse happesfingomüelinaasiks (ASM). See aine aitab lagundada (metaboliseerida) rasvainet, mida nimetatakse sfingomüeliiniks, mis leidub kehas igas rakus.
Kui ASM puudub või ei tööta korralikult, koguneb sfingomüeliin rakkude sees. See tapab teie rakud ja raskendab elundite nõuetekohast töötamist.
A-tüüp esineb kõigis rassides ja rahvustes. See on tavalisem Ashkenazi (Ida-Euroopa) juudi elanikkonnas.
Tüüp C tekib siis, kui keha ei suuda kolesterooli ja muid rasvu (lipiide) korralikult lagundada. See põhjustab maksa ja põrna liiga palju kolesterooli ning ajus liiga palju teisi lipiide. Tüüp C on kõige tavalisem hispaania päritolu Puerto Ricans.
Tüüp C1 on C-tüüpi variant. See hõlmab defekti, mis häirib kolesterooli liikumist ajurakkude vahel. Seda tüüpi on näha ainult Prantsuse Kanada elanikel Nova Scotias, Yarmouthi maakonnas.
Sümptomid
Sümptomid varieeruvad. Muud terviseseisundid võivad põhjustada sarnaseid sümptomeid. Haiguse varajased staadiumid võivad põhjustada vaid mõningaid sümptomeid. Isikul ei pruugi olla kõiki sümptomeid.
Tüüp A algab tavaliselt elu esimestel kuudel. Sümptomiteks võivad olla:
- Kõhupiirkonna turse 3 kuni 6 kuu jooksul
- Kirsipunane täpp silma tagaosas (võrkkestal)
- Söötmisraskused
- Varase motoorse oskuse kaotus (aja jooksul halveneb)
B-tüüpi sümptomid on tavaliselt kergemad. Neid esineb hilisel lapsepõlves või teismelistel. Väikestel lastel võib esineda kõhu turse. Peaaegu puudub aju ja närvisüsteemi kaasamine, näiteks motoorsete oskuste kaotus. Mõnedel lastel võib olla korduvaid hingamisteede infektsioone.
Tüübid C ja C1 mõjutavad tavaliselt kooliealisi lapsi. Siiski võib see esineda igal ajal varases lapsepõlves kuni täiskasvanueas. Sümptomiteks võivad olla:
- Raskuste liigutamise raskused, mis võivad põhjustada ebakindlat kõndimist, kohmakust, kõndimisprobleeme
- Laienenud põrn
- Suurenenud maks
- Kollatõbi sünnituse ajal (või vahetult pärast)
- Õppimisraskused ja intellektuaalne langus
- Krambid
- Ebatavaline, ebaregulaarne kõne
- Järsk lihaste toonuse vähenemine, mis võib põhjustada langust
- Värinad
- Silmade liigutamine üles ja alla
Eksamid ja testid
A- ja B-tüüpi diagnoosimiseks võib teha vere- või luuüdi testi. Katse võib öelda, kellel on haigus, kuid ei näita, kas olete kandja. DNA-teste saab teha A- ja B-tüüpi kandjate diagnoosimiseks.
C- ja D-tüüpi diagnoosimiseks tehakse tavaliselt naha biopsiat. Tervishoiuteenuse osutaja jälgib, kuidas naharakud kasvavad, liiguvad ja säilitavad kolesterooli. DNA-teste võib teha ka selleks, et otsida kahte sellist haigust põhjustavat geeni.
Muude testide hulka võivad kuuluda:
- Luuüdi aspiratsioon
- Maksa biopsia (tavaliselt ei ole vajalik)
- Slit-lamp silmade eksam
- Testid ASM-i taseme kontrollimiseks
Ravi
Praegu ei ole A-tüüpi efektiivne ravi.
B-tüüpi luuüdi siirdamist võib uurida. Teadlased uurivad jätkuvalt võimalikke ravimeetodeid, sealhulgas ensüümide asendamist ja geeniteraapiat.
C-tüüpi närvisüsteemi sümptomite korral on saadaval uus ravim, mida nimetatakse miglustaadiks.
Kõrge kolesteroolisisaldust võib ravida tervisliku, madala kolesteroolisisaldusega dieedi või ravimitega. Kuid uuringud ei näita, et need meetodid takistaksid haiguse süvenemist või muutust, kuidas rakud kolesterooli lagunevad. On olemas ravimeid, et kontrollida või leevendada paljusid sümptomeid, nagu näiteks lihaste toonuse ja krampide järsk kadu.
Tugirühmad
Need organisatsioonid võivad pakkuda Niemann-Picki haiguse kohta toetust ja rohkem teavet:
- Riiklik neuroloogiliste häirete ja insultide instituut - www.ninds.nih.gov/Disorders/All-Disorders/Niemann-Pick-Disease-Information-Page
- Riiklik Niemann-Pick haiguse sihtasutus - nnpdf.org
- Haruldaste häirete riiklik organisatsioon - rarediseases.org/rare-diseases/niemann-pick-disease-type-c
Outlook (prognoos)
A-tüüpi NPD on raske haigus. Tavaliselt põhjustab see surma 2. või 3. eluaasta jooksul.
B-tüüpi inimesed võivad elada hilisemas lapsepõlves või täiskasvanueas.
Laps, kellel on C-tüüpi märke enne 1. eluaastat, ei pruugi elada kooliealiseks. Need, kes pärast kooli sisenemist ilmutavad sümptomeid, võivad elada oma keskmisest-hilisest teismelist. Mõned võivad elada oma 20s.
Millal pöörduda arsti poole
Tehke oma teenusepakkujaga kohtumine, kui teil on perekonna Niemann-Picki haigus ja teil on plaanis lapsi. Soovitatav on geneetiline nõustamine ja sõeluuring.
Helista oma teenusepakkujalt, kui teie lapsel on selle haiguse sümptomid, sealhulgas:
- Arenguprobleemid
- Söötmise probleemid
- Kehv kaalutõus
Ärahoidmine
Kõik Niemann-Picki tüübid on autosoomsed retsessiivsed. See tähendab, et mõlemad vanemad on vedajad. Igal vanemal on 1 ebanormaalse geeni koopia ilma haiguse tunnustega.
Kui mõlemad vanemad on vedajad, on 25% tõenäosus, et nende lapsel on haigus ja 50% tõenäosus, et nende laps on vedaja.
Kandja tuvastamise testimine on võimalik ainult siis, kui on tuvastatud geneetiline defekt. A- ja B-tüüpi defekte on hästi uuritud. Nende Niemann-Picki vormide DNA testid on kättesaadavad.
Paljude C-tüüpi inimeste DNA-s on tuvastatud geneetilisi defekte. Võib olla võimalik diagnoosida ebanormaalset geeni kandvaid inimesi.
Mõned keskused pakuvad teste, et diagnoosida laps veel emakas.
Alternatiivsed nimed
NPD; Sfingomüelinaasi puudulikkus; Lipiidide säilitamise häire - Niemann-Picki haigus; Lüsosomaalse säilitamise haigus - Niemann-Pick
Pildid
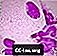
Niemann-Pick vahtrakud
Viited
Kliegman RM, Stanton BF, St. Geme JW, Schor NF. Lipiidide metabolismi defektid. In: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, eds. Nelsoni lastekirjanduse õpik. 20. toim. Philadelphia, PA: Elsevier; 2016: peatükk 86.
Patterson MC, Hendriksz CJ; NP-C suuniste töörühm jt. Soovitused C-tüüpi Niemann-Picki haiguse diagnoosimiseks ja raviks: uuendus. Mol Genet Metab. 106 (3): 330-344. PMID: 22572546 www.ncbi.nlm.nih.gov/pubmed/22572546.
Turnpenny PD, Ellard S. Ainevahetuse vigastused. In: Turnpenny PD, Ellard S, eds. Emery meditsiinilise geneetika elemendid. 15. ed. Philadelphia, PA: Elsevier; 2017: peatükk 18.
Läbivaatamise kuupäev 10/15/2018
Uuendatud: Anna C. Edens Hurst, MD, MS, meditsiini geneetika assistent, Alabama ülikool Birminghamis, Birminghamis, AL. VeriMedi tervishoiuvõrgustiku hinnang. Vaadatud ka David Zieve, MD, MHA, meditsiini direktor Brenda Conaway, toimetuse direktor ja A.D.A.M. Redigeerimismeeskond.